Population Dynamics
DEB theory has been set up for the purpose to specify (and understand) rules for how individuals interact with the environment that, in combination with models for environmental dynamics, determine population and ecosystem dynamics. Models for environmental dynamics should specify substrate dynamics (including transport and geochemical transformations), and the (micro)-climate (temperature and, in terrestrial environments, the dynamics of water). Given the DEB and environment models, population dynamics can be seen as an advanced book-keeping exercise which determines survival probabilities; ageing is rarely the cause of death of small-bodied species in natural populations, but still is important in population models to guarantee that individuals do not stay forever in the population. It sets a maximum to the memory of populations: population dynamics not only depends on the present state of the population (and environment), but also on its state in the (recent) past. This latter is usually taken into account by extending the states of individuals (e.g. including the ageing process and reserve). The book-keeping exercise is complex and, in practice, one is forced to apply a number of simplifications that raise problems by themselves. To structure the book-keeping, the (vector-valued) state of individuals is called the i-state (e.g. age, maturity, reserve, structure, reproduction buffer), and that of the population the p-state, i.e. the numbers of individuals with a given i-state for all possible values of i-states.
 Population dynamics is typically seen in the light of the ultimate aim of ecology: the understanding of ecosystem dynamics.
Ecosystems are a (large) set of interacting populations, where interaction is dominated by competition and predation,
while resource dynamics depends on nutrient recycling.
Predation-prey dynamics attracted a lot of attention in the literature, but its ecological significance in this context is limited
by the difficulty of selecting just two players from a complex interplay:
if predator dynamics depends on prey availability, why would this not apply to prey dynamics and that of the prey of the prey?
These models typically just count numbers of individuals in a population, ignoring properties of individuals,
which classify them as unstructured population models, to differentiate them from (physiologically) structured population models.
Models that assume homogeneous (well-mixed) space are called spatially implicit, to differentiate them from spatially explicit models.
Spatially implicit models are typically used to analyse model properties, not to produce realistic predictions.
The analysis can focus on transient behaviour, i.e. population-trajectories (functions of the p-state as functions of time),
or asympotic (i.e. very long term) behaviour.
The study of asympotic behaviour as function of parameter values is called bifurcation analysis.
Population dynamics is typically seen in the light of the ultimate aim of ecology: the understanding of ecosystem dynamics.
Ecosystems are a (large) set of interacting populations, where interaction is dominated by competition and predation,
while resource dynamics depends on nutrient recycling.
Predation-prey dynamics attracted a lot of attention in the literature, but its ecological significance in this context is limited
by the difficulty of selecting just two players from a complex interplay:
if predator dynamics depends on prey availability, why would this not apply to prey dynamics and that of the prey of the prey?
These models typically just count numbers of individuals in a population, ignoring properties of individuals,
which classify them as unstructured population models, to differentiate them from (physiologically) structured population models.
Models that assume homogeneous (well-mixed) space are called spatially implicit, to differentiate them from spatially explicit models.
Spatially implicit models are typically used to analyse model properties, not to produce realistic predictions.
The analysis can focus on transient behaviour, i.e. population-trajectories (functions of the p-state as functions of time),
or asympotic (i.e. very long term) behaviour.
The study of asympotic behaviour as function of parameter values is called bifurcation analysis.
Ecosystems have
- (primary) producers, which extract energy from light (plants, "algae", blue-green and sulphur bacteria) and/or minerals (some bacteria)
- consumers, which feed on primary producers, decomposers and consumers
- decomposers, which mineralize organic compounds (produced by primary producers and consumers) and close mineral recycling
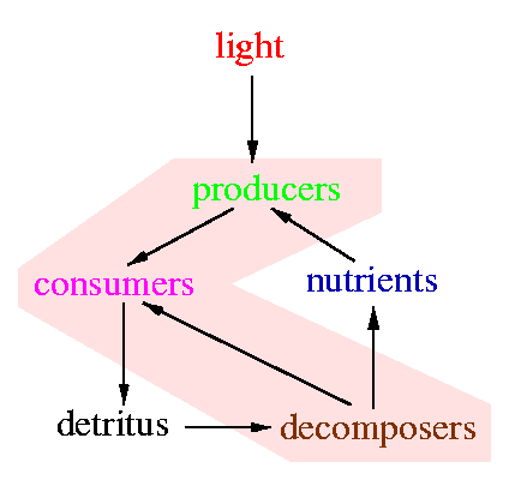 Animals are consumers, so they feed on other organisms, sometimes even their own species, forming food-chains and food-webs.
Parasites are a special type of consumers that are typically much smaller than their hosts and can affect their health.
Sublethal effects of parasites on their hosts are, like that of toxic compounds, incorporated via changes in parameter values,
such as maturity maintenance, which interacts with the defense system and affects the hazard rate.
Parasite population dynamics has similarities with that of most viruses, which have a degenerated i-state.
Epidemiology can be seen as a sub-field of population dynamics, with due attention for the health/illness dynamics of the (constant) host population.
Not all plants, "algae" and bacteria are primary producers, while almost all of them also consume organic compounds to some extent (mixotrophy).
Most fungi and bacteria specialize in the role of decomposers, frequently forming symbioses (e.g. mycorrhizas) with other organisms.
DEB models for micro-organisms (bacteria, "algae") have a number of reserves that equals the number of possibly limiting substrates (nutrients, light).
Plants additionally require multiple structures (root, shoot) and typically have more dynamic surface area to volume relationships, compared to animals.
Stoichiometric constraints on growth and reproduction complicate these models somewhat, but so far these DEB models fit data very well.
Since DEB models explicitly respect mass and energy conservation and specify product formation (faeces, N-waste, wood, metabolic water, etc),
they can be used to model syntrophic relationships, symbioses, food-chain and food-web dynamics, nutrient recycling, etc.
Since DEB theory formulates food acquisition in the framework of Synthesizing Units (which is Markovian stochastic by nature),
and the ageing process on the basis of hazard rates, while parameter values are individual-specific,
DEB-based population dynamics can naturally include stochasticity and has the ideal structure for Adaptive Dynamics (AD),
apart from the problem that AD rapidly becomes complex in the case of coupled traits.
Animals are consumers, so they feed on other organisms, sometimes even their own species, forming food-chains and food-webs.
Parasites are a special type of consumers that are typically much smaller than their hosts and can affect their health.
Sublethal effects of parasites on their hosts are, like that of toxic compounds, incorporated via changes in parameter values,
such as maturity maintenance, which interacts with the defense system and affects the hazard rate.
Parasite population dynamics has similarities with that of most viruses, which have a degenerated i-state.
Epidemiology can be seen as a sub-field of population dynamics, with due attention for the health/illness dynamics of the (constant) host population.
Not all plants, "algae" and bacteria are primary producers, while almost all of them also consume organic compounds to some extent (mixotrophy).
Most fungi and bacteria specialize in the role of decomposers, frequently forming symbioses (e.g. mycorrhizas) with other organisms.
DEB models for micro-organisms (bacteria, "algae") have a number of reserves that equals the number of possibly limiting substrates (nutrients, light).
Plants additionally require multiple structures (root, shoot) and typically have more dynamic surface area to volume relationships, compared to animals.
Stoichiometric constraints on growth and reproduction complicate these models somewhat, but so far these DEB models fit data very well.
Since DEB models explicitly respect mass and energy conservation and specify product formation (faeces, N-waste, wood, metabolic water, etc),
they can be used to model syntrophic relationships, symbioses, food-chain and food-web dynamics, nutrient recycling, etc.
Since DEB theory formulates food acquisition in the framework of Synthesizing Units (which is Markovian stochastic by nature),
and the ageing process on the basis of hazard rates, while parameter values are individual-specific,
DEB-based population dynamics can naturally include stochasticity and has the ideal structure for Adaptive Dynamics (AD),
apart from the problem that AD rapidly becomes complex in the case of coupled traits.
The following approaches, ordered from detailed to crude, can be used to follow the dynamics of populations, each with their own strong and weak properties.
- Individual-Based Models (IBMs): This approach follows each individual through time, possibly including spatial coordinates in inhomogeneous space. If space is discretized, and migration rules are specified, the models classify as cellular automata. The concept "individual" has been extended to "agent" to include social behaviour, leading to the term Agent-Based Models (ABMs). Spatial structure and interactions between individuals pose no problem for IBMs, although this can rapidly increase computational requirements. IBMs make use of the discrete nature of individuals, but a large number of individuals is a problem for this approach. For this reason, IBMs are rarely used for micro-organisms or other small organisms that can build up huge numbers of individuals. IBMs are too complex for mathematical analysis, restricting their use to simulate population size trajectories.
- Partial Differential Equations (PDEs): Individuals change their i-state continuously; PDEs follow the changes in the number of individuals with similar i-states: Physiologically Structured Population Models (PSPMs). The most popular methods to integrate pde's numerically to arrive at population-trajectories, discretize the values of (continuous) i-states and follow cohorts of similar individuals through time: the Escalator Boxcar Train (EBT) method. Finite difference methods discretize the i-states and follow changes in numbers of individuals in each cell. The PDE approach has been developed further to a coupled system of nonlinear renewal equations and differential delay equations for the analysis of asymptotic behaviour of PDEs. Spatial structure can be incorporated, using Lagrange-methods, but rapidly increases computational requirements. The number of individuals can be large without imposing problems for PDEs, but the discrete nature of individuals does. All individuals potentially contribute to the population-reproduction process. This is usually not a problem, but if food availability is very poor, such that no individual can produce a single offspring, the sum of such individuals still can in the PDE-approach (which is unrealistic). Direct interactions between individuals pose problems and can only be implemented via interaction with the environment. The dynamics of structured populations feeding on other structured populations is really complex. Considerable work has been invested for how to combine PDEs for the small-bodied (numerous) individuals with IBMs for the larger-bodied (less numerous) ones in multi-population models.
- Cohort Projection Models (CPMs): Like the EBT method, CPMs follow cohorts of similar individuals, assuming that environmental conditions synchronize reproduction events in the population and eggs of all individuals are the same. This allows to construct a map that projects the p-state at one reproduction event to the next. It can be seen as an intermediate between PDEs and integral projection models, which belong to the class of matrix models. The number of different cohorts equals the maximum age devided by the interval between reproduction events. Since this number is much less, relative to the EBT method, CPMs require much less computation time.
- Matrix Models (MMs): The population is sub-divided in a limited number of classes corresponding to the states of individuals, similar to PDEs. Time is also discretised. A state vector counts the number of individuals in each class, and a matrix of transition probabilities specifies possible changes in the state of individuals. The set-up is very similar to discrete-time, discrete-state Markov-chain, the continuous-time version is called Markov-process. The MM-approach has become popular in ecology, since it is computationally simple and fast. Where PDEs still follow individuals, MMs only deal with changes in states, not how long an individual has been in that state, for instance. Integral Projection Models (IPMs) avoid discrete state-classes, linking to discrete-time, continuous-state Markov-chains. This can be implemented with standard matrix software. Both MMs and IPMs classify as Demographic Models, since changes in i-state depend on time only, not on other i-states. This can present a serious drawback for DEB applications. State-structured models always reduce to age-structured models in constant environments. If food densities fluctuate, however, this is no longer the case and this mismatch can have strong effects: MM-generated population-trajectories can deviate substantially from the ones implied by DEB models, due to the reduced book-keeping. This reduced book-keeping also complicates explicit mass and energy conservation, which makes it hard to model nutrient recycling.
- Ordinary Differential Equations (ODEs): Depending on the model for individuals, PDEs for populations can sometimes be reduced to a finite set of ODEs that include particular changes in i-states, sharing the advantages of PDEs over ODEs. This is called the Linear Chain Trick (LCT), where not only numbers of individuals, but also total length (of all individuals combined), surface area and volumes are followed. It can only be applied to DEB models using additional approximations, such as assuming equal reserve density for all individuals, while DEB theory actually implies that small-bodied individuals follow fluctuations in food density faster than big-bodied ones. The simplification is both the strength and the weakness of ODEs, and still attractive if changes in states of individuals have little effect on population dynamics. While for micro-organisms this might hold by rough first approximation, for other organisms this might not be realistic: the contribution of neonates to reproduction easily dominates the increase in numbers because of the interest-on-interest principle. Energy and mass conservation can only be respected in a very crude way with the ode-approach, although event-driven stochastic ODEs have been formulated that respect mass conservation. Population models in the past (before 1980 say) ignore the state of individuals and just follow total mass or number of individuals. Since PDEs can been seen as an infinitely large set of coupled ODEs, it is easy to see the substantial simplification that is obtained by ODEs, compared to PDEs. This simplicity allows the partitionning of space into cells, each with its own population and immigration/emigration rules: the meta-population models.
- Pseudo Steady States (PSSs): Given a constant food level, populations eventially grow exponentially and (generally) have an associated stable size (and age) distribution. The rate of population growth can be found by solving the characteristic equation. DEB models specify all mass that is consumed (food, dioxygen) and produced (feaces, minerals, dead bodies) by the population. The crudest of all approaches assumes that this still applies when food density is (slowly) changing in time and reduces population dynamics to a macrochemical reaction equation with time-varying coefficients. The method can be close to accurate provided that changes in food density are slow enough (in relation to growth and reproduction) and can be valuable in the context of biotechnology and ecosystem dynamics.
From AmP entries to animal population dynamics
The code for population dynamics is part of DEBtool, in subdirectory popDyn. These functions import DEB parameter values for any species directly from the AmP collection; DEBtool itself has no parameter values. If this is the first time you are using popDyn, make sure that you have the most recent version of DEBtool_M, and make sure to reset the matlab path to DEBtool_M, including subdirectories.The step from AmP entries to population dynamics needs the following
- extra parameter(s) for the specification of the reproduction buffer handling rules (spawning), respecting the ecotype for reproduction, gender and possibly migration and embryo environment. DEB theory has a number of frequently occuring reproduction buffer handling rules.
- extra parameters for the hazard rate given the i-state.
Basic causes of death are
- aging: part of all AmP entries (Weibull aging acceleration ha and Gompertz stress coefficient sG)
- starvation: part of DEB theory, but not applied in AmP. The extra parameters are the maximum shrinking fraction of structure δX, the rejuvenation rate kJ′, and the rejuvenation hazard hJ.
- accidental (background): stage-specific, but independent of the i-state.
- thinning: not part of DEB theory. If the hazard rate equals ⅔r for isomorphs, or r for V1-morphs, where r is the specific growth rate of the structure of a DEB-individual, the total feeding rate of a cohort of individuals does not change during growth at constant food, since the implied increase in feeding by cohort members is compensated by a decrease in numbers. This can be used to avoid that a very large number of tiny neonates from a single mother turns population dynamics unrealistic if they survive too long in the model. Some species don't survive thinning in the long run.
- specification of the founding population (p-state at time zero)
- prescribed trajectories for temperature and a specification of food dynamics
To accomodate the energy cost of male production in the case of gonochoric species in a DEB-context, reproduction efficiency is halved on the assumption that the sex ratio is 1:1, since any difference between sexes typically develop slowly during ontogeny. Many entries have parameters for males that deviate from that of females, which can be taken into account in a straightforeward way.
All DEBtool support for population dynamics offers two ways to specify species-parameters:
- via a character string with the name of an entry, where the parameters will be read from
allStat.mat, which needs to be downloaded as part ofAmPdatain theCOLLECTIONdropdown in the toolbar above this page. - via a string of 3 cells from the
results_my_pet.matfile, which has the structures metaData, metaPar and par. This.mat-file needs to be downloaded from the entries-directory of AmP and, if loaded into Matlab, these three structures are created, which you can modify before use.
We now discuss how to apply the various approaches to any AmP entry.
No interactions between individuals
Individuals do not interact in PSSs, since food density is assumed to be constant; methods that include interaction are described below. Temperature is also taken to be constant; starvation and the founding population are not relevant since only pseudo steady states are considered.Pseudo Steady States (PSSs) in AmP
For PSSs, as presently included in AmP, the buffer handling rule is to produce offspring as soon as the reproduction buffer allows.All entries have a page "Population traits", where the PSS-approach has been worked out in presence and absence of thinning. Apart from a very high food level, a low one for which the population growth rate is zero is shown (of females), as well as an intermediatiary scaled functional response, in the case where males and females have the same parameters. If the parameters differ between sexes, the latter is suppressed to avoid too large tables. If a table has NaN's (Not-a-Number), this typically means that the species does not survive thinning. The scaled functional response for which the specific population growth rate is zero relates to the female parameters; males might not reach puberty at this food level, with NaN's as result. Non-smooth looking survivor curves originate from numerical problems (and are not model implications), typically caused by puberty being reached only very close to the ultimate size.
The population traits include the yield coefficients of the macro-chemical reaction equation:
X + YOX O →
YPX P + YVX V + YVX† V† + YEX E +
YEX† E† + YCX C + YHX H + YNX N + μTX
for food X, dioxygen O, faeces P, living structure V, dead structure V†, living reserve E, dead reserve E†,
carbon dioxide C, N-waste N and heat μTX.
The yield coefficients Y are ratios of fluxes and depend on stable age distributions, which are presented as graphs.
Dioxygen is assumed to be not-limiting.
The water flux includes only metabolic water, not evaporating water (since this needs to be compensated by drinking, which is not included in PSSs).
The default dry-wet weight ratios are specified in get_d_V,
the N-waste in get_N_waste, for each taxon.
The default values for the chemical indices and chemical potentials are given in addchem.
All default values can be overwritten by the user.
The yield coeffients for the minerals (C, H, O ,N) follow from those for the organics (X, V+V†, E+E†, P) via the conservation of chemical elements. The mean feeding rate is given by JX = f {JXm} L2, where L2 is the mean squared structural length in the population, and f the scaled functional response. Likewise, the mean defacation rate is given by JP = yPX JX. The mean production rate of living structure is found from JV = rN [MV] L3, where rN is the specific population growth rate, and the mean production rate of living reserve is JE = JV f [MEm]/[MV], if we ignore the contribution of yolk in eggs. The mean production rate of dead structure is JV† = [MV] h L3, where h L3 is the mean product of the hazard rate and cubed structural length in the population. The mean production rate of dead reserve JE† = JV† f [MEm]/[MV]. Finally, the organic yields are Y*X = J*/JX, where * is P, V, V†, E and E†.
The fact that most species reproduce in clutches (or litters), typically in response to a cyclically behaving environment, rather than one-by-one, is difficult to accomodate in the pss-approach, which assumes that the environment stays constant (in terms of temperature and food). Clutches reduce the population growth rate, due to the interest-upon-interest principle, where the very first young contributes most since it is first ready to reproduce itself, while clutches require longer accumulation time of the reproduction buffer. For semelparous species, which produce a single clutch of, say, N eggs, at the end of their life at age am, while they have probability Sm to reach that point, the population growth rate becomes log(Sm N)/ am.
The species-subdirectories of the directory entries_web,
where the population-trait-pages are parked, also have a file popStat.mat,
i.e. a structure that has all numbers shown on these pages.
The fuction write_popStat collects these structures, via the web, into one single structure for all species (similar to how allStat.mat
collects DEB parameters and other traits for all species).
This can be handy for comparison of population traits amoung species.
The population-trait-page can also be generated in the context of paramater estimation for a species that is not yet in the collection, by choosing
estim_options('results_output', 6) in run_my_pet.m,
i.e. one of the 4 files that are required to estimate parameters from data in the AmP-system.
The pages have a default-setting of background hazards equal to zero.
Independent from the estimation context, the user can choose different values for background hazards with AmPtool function
prt_my_pet_pop,
which also holds for other values of temperature and scaled functional response.
In that case the parameters are extracted from allStat.mat.
To change the stage-specific background hazard rates, temperature and scaled function response for an existing species in the collection and re-compute
the population-trait-page locally:
- specify the background hazards in d-1, e.g. for embryo, juvenile and adult in the std-model
h_B = [1e-4; 1e-5; 2e-4].
The possible hazards for that model can be read from the population-trait-page; default is zero for all background hazards. - specify the body temperature in Kelvin, e.g.
T = C2K(22.3). - specify the scaled functional response, e.g.
f = 0.85. - run
prt_my_pet_pop(my_pet, h_B, T, f), where the last 3 inputs are optional, and inputmy_petthe name of your species. - open the file
my_pet_pop.htmlthat has been written in your browser.
- first download the
add_my_pet/entries/my_pet/results_my_pet.mat, withmy_petreplaced by the name of an AmP species (2 ×), - load it in Matlab with
load results_my_pet, which creates the structuresmetaData,metaPar,par(withmy_petreplaced by the name of an AmP species). - modify values in the par-structure, e.g.
par.p_M = 1422and - run
prt_my_pet_pop({metaData, metaPar, par}).
Interactions between individuals
Individuals interact via competition for food; more advanced forms of interaction are beyond the present implementation. Yet, such interactions can be crucial for population dynamics. If space is homogeneous and feeding rate only depends on food density, competitive exclusion results, implying loss of species diversity. With only a single extra parameter, social interaction can be included, such as spelled out in section 7.2.4 of DEB3, allowing for the co-existence of many species on a single food resource. This is because feeding now not only depends on food availability, but also on population density.CPMs are coded within DEBtool, but for EBTs and IBMs, DEBtool just provides a Matlab-shell around external code, to handle in- and output, as well as extracts parameter values from the AmP collection for a specified species, which follows a particular model. The general idea is that DEBtool writes input-files with model-specification and parameter-values for this external code, runs the code in Windows Powershell or the Mac-equivalent and reads the output files produced by this code, for presentation. The hope is that once it is clear how to do this, the route to more advanced applications is open for the user.
DEBtool, in combination with AmP results, focuses on the first steps in the direction of ecosystem dynamics: population dynamics in a generalized well-stirred reactor, and allow the comparison of several methods to follow population dynamics. Well-known generalized reactors are fed-batch reactors, which have no output, and chemostats, where the hazard for substrate input and all background hazards are equal. Such a reactor has a substrate (nutrients, light, food) input, and an output of substrates, individuals and their products, each at a specified rate. The task of population dynamics is here to evaluate the dynamics of the contents of the reactor, respecting mass and energy conservation.
In a generalized reactor, food input and the background hazard rate for food and for each stage of individuals can be set independently.
Thinning can optionally be applied, but aging is allways included.
The change in food density X is
dX/dt = JXI/VX - hX * X - {JXAm} * X/(K + X) * ∑i Ni * Li2
where JXI is food input, VX reactor volume, hX hazard rate for food, {JXAm} maximum specific ingestion rate,
K half-saturation constant, Ni number of individuals per volume in cohort i, Li structural length in cohort i.
The summation is over all cohorts, excluding embryos (because they do not eat).
The cohorts experience background and aging hazards and, optionally, thinning.
If the background hazards equal the hazard for food, the reactor behaves as a chemostat; if they are all equal to zero, it behaves as a fed-batch reactor.
The dimension volume in reactor volume VX, can be replaced by surface area, if that also occurs in food density X, half saturation constant K and specific searching rate {Fm}, which is the DEB core-parameter behind the half-saturation constant. This typically applies to most terrestrial species, and the larger-bodied aquatic ones: they live in 2D, rather than 3D, but some live in the twilight zone between 2D and 3D.
Semi Stuctured Model (SSMs) in AmP
The Semi Structured Model delineates the life stages embryo, juvenile and adult, but treats individuals within these three cohorts as identical. The transition rates are based on the stable age distribution, which is assumed to apply for each point in time, despited changes in food density. Reserve is treated as being in instantaneous equilibrium and the maternal effect rule is not respected. The model is specified in the Comments for Section 9.2.2.5. Function SSM computes the trajectories of densities of food (scaled), total number, length, surface, volume and weight, e.g.txNL23W = SSM('Torpedo_marmorata');.
The behaviour of this model differs substantially from more detailed ones, due to the distribution of residence times of individuals in each stage.
Cohort Projection Models (CPMs) in AmP
For CPMs, as presently included in DEBtool, the buffer handling rule is to produce offspring at fixed moment in a year, each year (the period can be set differently). This is not a likely scenario for all species and, for this reason, not implemented for hep and hex models. Contrary to PSSs, all freshly layed eggs, or newborn foetuses, are assumed to be identical, so deviate from DEBs maternal effect rule. Food input and temperature trajectories are prescribed (constant or seasonally varying using knots in a spline), while food density results from competition and can change in time. All control parameters are set by default, which the user can overwrite. Like for PSSs, two input routes for species-parameters are implemented in CPM. The simplest use ofCPM is via allStat with e.g. [txNL23W, M_N, M_L, M_L2, M_L3, M_W, NL23Wt] = CPM('Torpedo_marmorata');.
More flexible is via a cell-string, rather than the name of an entry:
- first download
add_my_pet/entries/my_pet/results_my_pet.mat, withmy_petreplaced by the name of an AmP species (2 ×), - load it in Matlab with
load results_my_pet, which creates the structuresmetaData,metaPar,par(withmy_petreplaced by the name of that AmP species). - optionally modify values in the par-structure, e.g.
par.del_X = 0.5and - run
[txNL23W, M_N, M_L, M_L2, M_L3, M_W, NL23Wt] = CPM({metaData, metaPar, par});.
CPM generates 7 figures and 2 html-pages (which are opened), one with species-properties (no direct connection with CPM but shows implications of
possible changes in parameter setting via cell-string input) and one with all CPM-parameter settings.
The first output txNL23W gives the scaled food density, x = X/K, and the total number N and sums of lengths to the power 1, 2 and 3
and total weight W as functions of time t.
The last output NL23Wt of CPM is an array with the values of N, L, L^2, L^3 and W for the various cohorts at the last simulation time point.
The outputs MN till MW of CPM are the projection maps in N(t+tR)=MN*N(t) and
W(t+tR)=MW*W(t), where N is the vector of numbers of individuals in the various cohorts and W the total wet weights, both as densities,
with tR the time between reproduction events.
The maps are computed from the time-trajectories and are only reliable if the simulation is over enough reproduction events.
This makes the link to matrix models (MMs), see website comadre.
Many MMs applications treat the time interval between subsequent steps as incremental (i.e. changes in i-states are assumed to be small),
but in CPMs it is the reproduction interval, typically a year.
IPMs generally don't treat the time interval as incremental.
The required running time not only depends on the selected number of reproduction events, but also on parameter settings: Integration with rapid changes is more computanionally intensive. Species that require high food levels to reach puberty are difficult to maintain is this type of reactor. CPMs are described in KooiKooy2020.
The Matlab-code that specifies the various DEB models of AmP is located at DEBtool_M/popDyn/ in functions called dCPM***.
Escalator Boxcar Trains (EBTs) in AmP
EBTs are implemented in DEBtool via EBTtool of André de Roos. The code itself is already in DEBtool, and differs from the original code, e.g. by omitting the graphical shell. It is written in C and needs compilation, which is done by Matlab function EBT in Windows 10 PowerShell (and also runs on Mac). This needs to be prepared, however, by installing a C-compiler with the namegcc.exe, e.g. the
MinGW for the GNU Compiler Collection for Windows, see YouTube for installation instructions,
which also shows how to set a path to this compiler.
EBTtool can handle a wider set of applications than implemented in DEBtool.
The only aim here is to fascilitate the initial use of EBT in the context of AmP, showing how the parameters of AmP can be extracted and the models specified.
Like CPMs, all freshly layed eggs, or newborn foetuses, are assumed to be identical, so deviate from DEBs maternal effect rule. Contrary the CPM, the EBT impementation in DEBtool does not assume synchronized reproduction: offspring is produced as soon as the reproduction buffer allows. Temperature and food input can, like with CPMs, chosen to be constant or specified via knots of a spline, and not necessarily be cyclic. Also like CPMs, the species-parameter input can be via an entry-name or, more flexible, via a cell-string.
Running EBT is exactly like CPM, but the time in the knots for food supply and temperature are not in scaled time, but absolute time,
spanning the full range from zero to maximum simulation time.
So txNL23W_EBT = EBT('Daphnia_magna'); gives results that can be compared with txNL23W_CPM = CPM('Daphnia_magna');.
Again it is possible to run EBT via EBT({metaData, metaPar, par}), which allows the user to modify parameter settings before use.
Contrary to CMP, function EBT does output the linear maps, since synchronized periodic reproduction is not assumed.
The C-code that specifies the 11 different (but related) DEB models of AmP is located at DEBtool_M/popDyn/EBTtool/deb. The user can modify this code to include particular 'details', such as other reproduction buffer handling rules.
Individual-Based Models (IBMs) in AmP
IBMs are implemented in DEBtool via NetLogo. If not already installed, first download Netlogo (version 6.2.0 was tested, but 6.3.0 gave problems) and set a path to it in Windows,Mac or Unix as well as to java.exe.The maternal effect that reserve density at birth equals that of the mother at egg-laying is implemented in IBM. The gender is set at creation of each new individual; males might differ from females by {pAm} and EHp. Three buffer-handling rules are implemented: spawn if (1) the buffer has enough for an egg, (2) the accumulation time equals the incubation period or, (3) a specified period. Temperature and food input can, like with CPMs and EBTs, chosen to be constant or specified via a spline; the absicca of the knots are in real time, not scaled time. Also like CPMs and EBTs, the species-parameter input can be via an entry-name or, more flexible, via a cell-string.
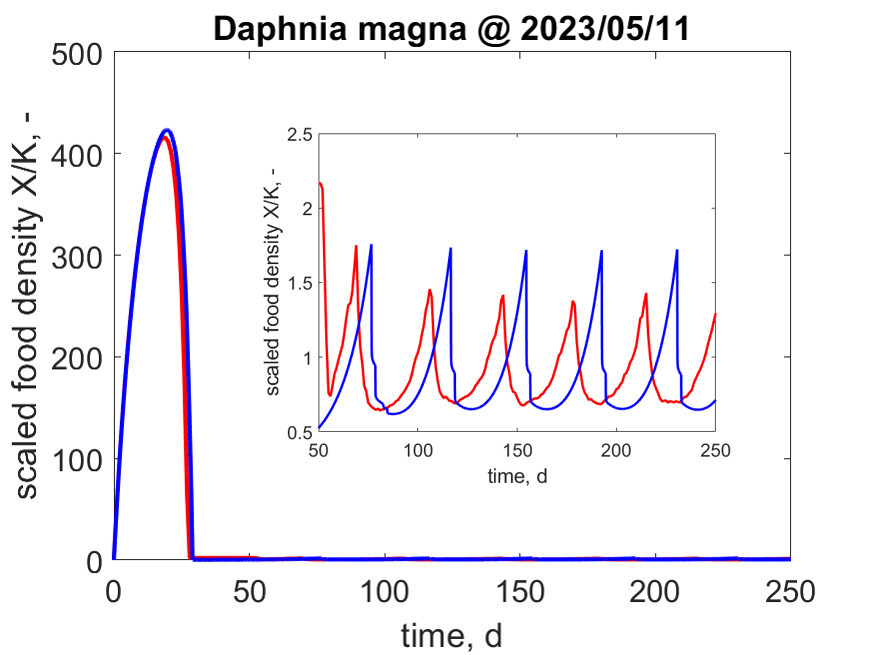

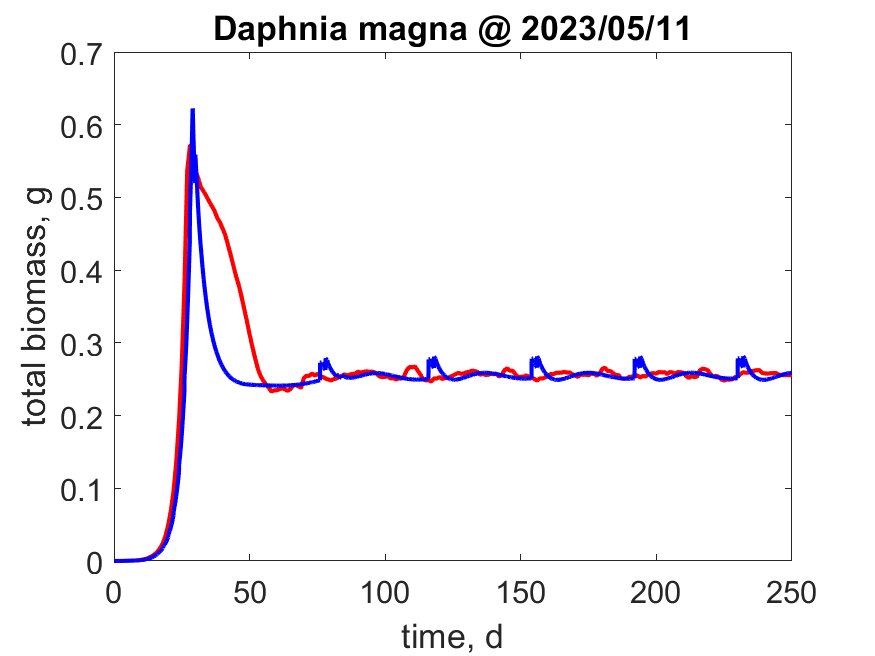 Running
Running IBM is exactly like CPM and EBT.
So txNL23W_IBM = IBM('Daphnia_magna'); gives results that can be compared with txNL23W_CPM = CPM('Daphnia_magna');
and txNL23W_EBT = EBT('Daphnia magna');.
Again it is possible to run IBM via IBM({metaData, metaPar, par}), which allows the user to modify parameter settings before use.
IBM terminates if t_max is exceeded or the number of individuals becomes 0 or larger than 15000. EBT can be used to find reactor parameter settings that produce a reasonable number of individuals; its running time does not depend on numbers.
Remark: work in progress; will work soon, please be patient.
Near-future project: IBMs via C++ code that was written by Chiara Acolla. It needs to compile C++-code, see, e.g. the MinGW for the GNU Compiler Collection for Windows, and YouTube for installation instructions.